
Debates over the contribution of genetics to differences among populations have a long and contentious history. We have known for a long time that nearly all traits are partially heritable, meaning that genetic differences are associated with differences in phenotypes within populations (as are differences in environment). However, if a trait is highly heritable within a population, it doesn’t follow that differences between populations are due to genetics– environmental and cultural differences could instead be the primary driver of between-population differences.
Recently the field of genetics has made huge progress in identifying regions of the genome (single nucleotide polymorphisms, SNPs) that are associated with differences among individuals within a population, using genome-wide association studies (GWAS). GWAS studies have found SNPs associated with a dizzying array of traits, including behavioural traits, and sophisticated methods for estimating heritabilities have also emerged. The success of GWAS seems to suggest that we’ll soon be able to settle debates about whether behavioural differences among populations are driven in part by genetics. However, answering this question is a lot more complicated than it seems at first glance. In this blog post I’ll talk through some of the complications, including how gene-by-environment interactions and correlations among SNPs make it difficult to use polygenic scores to understand differences among populations.
Some of these complications are perhaps best illustrated with a toy example. Say we perform a GWAS of the amount of tea that individuals in the UK drink (e.g. in the UK Biobank). On the basis of this tea GWAS, someone (let’s call him Bob) could claim that we could learn about France-UK differences in tea consumption by just counting up the average number of alleles for tea preference that individuals in the UK and France carry. If the British, overall, are more likely to have alleles that increase tea consumption than French people, then Bob might say that we have demonstrated that the difference between French and UK people’s preference for tea is in part genetic. Bob would assure us that these alleles are polymorphic in both countries, and that both environment and culture plays a role. He would further reassure us that there’ll be an overlapping distribution of tea drinking preferences in both countries, so he’s not saying that all British people drink more tea for genetic reasons. He’ll tell us he’s simply interested in showing that the average difference in tea consumption is partly genetic.
At face value, Bob’s argument seems scientifically sound; If there are alleles for tea preference to determine whether a British people’s love of a good cuppa tea is genetic, Bob just need to count these alleles up and compare them to the average allele counts in France. Adding up these tea preference alleles for individuals is one way of calculating an individual’s “polygenic score”. Polygenic scores are predictions of people’s traits computed from genotype data. There are several ways of calculating polygenic scores, and they have a range of potential uses. For example, people have done GWAS for risk of heart disease, and the resulting scores may offer a way forward in enabling preventive care. Currently, these polygenic scores often do not explain a lot of the variation in traits, but the size of studies is increasing, and predictions based on polygenic scores will become more accurate (within populations).
Now polygenic scores constructed using GWAS information from a single populations are expected to differ among populations. The allele frequency at every locus will vary among populations because of genetic drift, the compounding of chance variation in allele frequencies across generations, leads allele frequencies among populations to diverge over time. (If natural selection acts on the locus differently in the two populations, it also cause allele frequencies to differ.) Since a polygenic score is just a weighted sum of allele frequencies, it will also vary among populations. Importantly, however, that does not imply that genetics must contribute to an observed difference in phenotype among populations. It could be the case that French people tend to have higher polygenic scores for tea-consumption than the British, but that this genetic predisposition is hidden or counter-acted by cultural influences. For example, perhaps British people on average find bitter (tannin) tastes slightly less palatable than French people, but this influence is overridden by the culture of tea drinking in the UK.
Even beyond the fact that environment and culture can overwhelm the influence of genetics, there’s another, deeper problem: polygenic scores are not strong statements about differences in the contribution of genetics to phenotypic variation among populations. The issue is that GWAS studies do not point to specific alleles FOR tea preferences, only to alleles that happen to be associated with tea preference in the current set of environments experienced by people in the UK Biobank. Similarly, as geneticists, we talk about height alleles. But these are not alleles FOR height, but simply alleles that are associated with differences in height within a population. There’s no guarantee that alleles mapped within populations will affect the trait in the same way in other populations and environments, nor (even if they do) that they will explain differences between populations.
Complex traits are just that—complex. Most traits are incredibly polygenic, likely involving tens of thousands of loci. These loci will act via a vast number of pathways, mediated by interactions with many environmental and cultural factors. Some of our tea-GWAS SNPs may well be enriched near olfactory receptors and genes expressed in relevant parts of brain, and some may overlap with SNPs associated with caffeine sensitivity. But the majority may not, they will often fall near genes with no simple connection to our trait. The rare cases where we can confidently make a specific causal connection to a gene and through a causal pathway all the way to phenotype may explain so little of the variance that, while they may provide important clues to biology, they often won’t allow us to state a general causal mechanism that explains a lot of the variance. In saying this, I’m not anti-GWAS. We have learned a lot of new biology from GWAS, and doubtless will learn a lot more over the coming decades. But they are far from a complete solution to understanding the causes of variation, especially variation among populations. Let’s see why.
Gene-by-environment interactions (G x E)
The effect of an allele on any given phenotype is always in the context of a particular set of environments. This issue is not new: debates over the meaning of heritability and the genetics in the context of environmental variation stretch back to the dawn of quantitative genetics (see, e.g., the debate between Hogben and Fisher). These issues are particularly difficult in humans, though, as we cannot raise humans in laboratory environments or randomized environments. Our behavioural, cultural and societal practices will influence the ways in which genetic variants impact phenotypic variation.
For example, there are cultural differences between the UK and France in whether milk is taken with tea, in the types and quality of tea drunk, and in the prominence of coffee. What role do parents, and older siblings, play in an adult’s choice of beverage, which shape indirect genetic effects, and how does these differ between countries? Presumably all of these differences, and many others, could mean that the genetic basis of tea drinking will differ between France and the UK. Therefore, the loci that influence tea drinking in the UK could be somewhat different from those underlying differences in tea drinking in France.
Suppose after our GWAS for tea drinking in the UK and France, we found that the genetic basis of the trait within both countries was correlated. What would be a high enough correlation to constitute evidence of a genetic difference in phenotypic preferences between countries? Moreover, even if the polygenic score explained a lot of the variance within each country, it may not explain much of the difference between the countries. As one example: maybe if people who care about their weight more are more likely to drink tea (e.g., as compared to soda), then alleles that are correlated with BMI in the UK Biobank will be alleles that predispose you to tea drinking. These loci may be reliably associated with BMI and tea drinking in both the UK and France. Yet a difference in the frequency of loci associated with BMI between the UK and France would not imply that differences in tea drinking preferences among countries result from genetics. Suppose for example that an individual’s preference for tea is not influenced by their absolute BMI, but rather by their relative BMI within a country, because of how they feel about their weight relative to people they regularly encounter. In this scenario, a polygenic score could be predictive of individual’s phenotypes within multiple countries but have little predictive power in explaining differences among those countries.
Without a thorough understanding of the casual biological and cultural mechanisms by which GWAS SNPs interact with the range of environments encountered by individuals, it may be hard to rule out GxE as a serious confounder of inferences of polygenic scores across populations.
We don’t have the functional genetic markers.
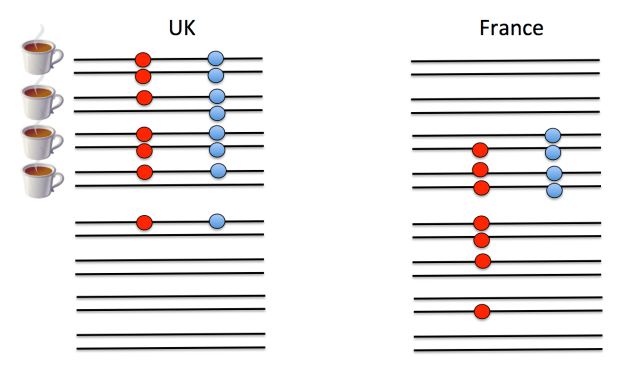
A second major hurdle that we face in understanding polygenic scores is that we do not know the loci that are functionally important for trait variation, only loci that are statistical proxies for them—sometimes called tag SNPs—that will be nearby in the genome. (Technically the SNPs used to construct polygenic scores are in linkage disequilbrium with the functional loci—meaning that genotypes at the tag SNP are correlated with genotypes at the functional locus—but unlikely to be the functional loci themselves.) To understand this point, look at the example below. On the left is a cartoon of people from the UK. Each person has two chromosomes (horizontal black lines) and in this small stretch of the genome there are two loci (red and blue SNPs), the alleles of which are indicated by the presence/absence of a filled circle. Whether an individual drinks a lot of tea is indicated by the tea cup next to the individual. Both of the filled circle alleles appear to be associated with tea drinking. (Obviously this sample size is laughably small, but you get the point.) However, only one of them is the functional SNP predisposing people toward tea drinking; the other SNP just happens to be associated because the mutation there arose at a similar point in history on the same genetic background. If we guess that the blue allele is the functional one, we would predict that French people have a slightly weaker preference to tea on the basis of this allele. But if we guess the red allele is the functional one, we would predict that the UK and France have very similar tea drinking habits on the basis of this locus.
What’s happened here is that the correlation between the alleles at the two loci have changed due to different histories of recombination and genetic drift. Now such a strong change in the correlation of loci is unlikely between two countries, such as Britain and France, that share so much of their genetic history. However, it is a serious problem when comparing populations that have been more distant from each other for a longer period of time. The fact that the correlation between any two SNPs changes over evolutionary time is a major reason why polygenic scores lose predictive ability as we move to populations that have been isolated from each other for more of their history. Even for closely related populations, it may be a problem when we consider that the many weak GWAS signals that likely much of the heritability for typical traits, as these associations may be due to collections of loosely linked SNPs. One way forward would be to perform GWAS in multiple populations and try to narrow down the actual functional SNPs, but again, this is no small undertaking.
A second, more subtle force can decrease the predictive validity of polygenic scores. Assortative mating among individuals can drive rapid changes in the SNPs associated with a trait. For example, if people who drink more tea tend to have children with taller people, this pattern of assortative mating can cause greater height and tea drinking to become associated (formally can lead to a genetic correlation). In other words, height-increasing alleles will be associated with tea drinking because the offspring of tea-drinking/tall couples will have alleles associated with both tea drinking and height. Even after assortative mating has stopped, these effects can persist for a few generations, making them potentially hard to rule out. Such associations need not hold in other populations, however, if they do not have similar patterns assortative mating. Therefore, sets of loci that contribute to trait variation via genetic correlations may change rapidly across environments or populations due to shifts in assortative mating.
We will not map within a single population all of the alleles influencing trait differences among populations.
GWAS have the highest power to map alleles that are present at intermediate frequency in the GWAS population (all else being equal). The functional variants contributing to a trait will differ in frequency among populations due to genetic drift and selection; therefore, GWAS will miss many of the loci contributing to phenotypic variation in other populations. This may not be much of a problem for comparing the UK and French population, as allele frequencies are very similar in the two countries. However, it’s potentially a much bigger problem in comparing more distant populations.
An example of the complexity of the ways in which different variants contribute to a trait in different areas of the world is the genetics of skin pigmentation. The variants that were mapped within European populations, though important in Europe, explain little of the variation in skin pigmentation worldwide. Even variants that explain the lighter European skin pigmentation do not explain the lighter skin pigmentation in East Asians (e.g. see here and here). Work from Sarah Tishkoff and Brenna Henn‘s labs has demonstrated that a number of loci important for explaining skin-pigmentation variation world-wide were missed by studies focused on non-African populations. A big part of the story was missing until variation within Africa was explored, with undoubtedly much more to uncover about this trait from GWAS in many populations. Furthermore, our understanding of the evolutionary history of skin-pigmentation in Europe has been majorly revised by ancient DNA. This history of major shifts in our understanding of the genetics and evolutionary history of skin pigmentation suggests that bold claims about other traits, based on incomplete evidence, may well not stand the test of time.
In the coming decade, we will likely uncover a surprising amount of heterogeneity in the alleles controlling trait variation world-wide. Based on genetic drift alone, we should expect as much: the alleles that explain most variance in populations of European ancestry will not be the same alleles in East Asia as allele frequencies drift over time. Also as a result of allele frequency change at many loci, across populations, epistatic relationships among loci may also change in unpredictable ways, confounding cross-population predictions.
These problems of different alleles contributing to traits in different populations will be compounded for traits subject to natural selection (as well as genetic drift). Whether traits are subject to stabilizing selection or directional selection (shared or divergent), selection will drive more rapid turnover in the loci contributing to trait variation among populations.
Again, one can hope to address these issues by performing GWAS in multiple worldwide populations, but we should expect to have a European-biased view of genetic variation for some time to come, simply because of the size of the studies in these populations dwarfs those done elsewhere.
Conclusion
Undoubtedly the coming decades of human genomics will see breakthroughs in the identification of functional loci, the size of GWAS performed world-wide, and in the statistical methodologies used to understand trait variation. There is also no doubt that we will come to understand much more about human variation. However, our ability to perform GWAS to identify loci underlying variation in traits among individuals vastly outstrips our ability to understand the causal mechanisms underlying these differences. In many cases, genetic contributions may not be separable from environmental and cultural differences. Certainly making a case for the relative importance of genetics in explaining among-population differences will involve a lot more work than simply counting up the number of tea preference alleles in populations and seeing how the averages differ.
These complications notwithstanding, I suspect that over the next decade, we are going to see a lot of partial results and incomplete (and in some cases initially downright incorrect) stories about the genetics of among-population variation in traits. For example, we now think we know something about the evolution of polygenic height scores among European populations. Results in hand allow us to demonstrate that natural selection has likely driven the higher polygenic height scores of Northern Europeans compared to Southern Europeans (Turchin et al, Berg and Coop, Robinson et al Mathieson et al, Berg, Zhang, & Coop). But they do not convincingly demonstrate that among-population differences in height in Europe are genetic (for all the reasons outlined above; for more, see here). Furthermore, our understanding of height genetics drops off quickly as we move away from Europe: we are even further away from understanding height differences among populations across Eurasia, and European-GWAS polygenic height predictions are positively misleading when applied to African populations. The complexity of such partial results reflects our uncertainty about the genetics of height–and that’s for height, an easily measured and well-studied trait. Applied to other and more fraught traits, this patchy understanding of the contribution of genetics to phenotypic differences will be fertile ground for misleading claims.
Finally, there is a more fundamental disconnect between talk of polygenic scores and what some people seem to think they might learn from this kind of research. Even if we could attribute some proportion of the phenotypic difference to a difference in polygenic score, on a deeper level, it is not even clear whether such a result really answers the question that an average person means to ask when they ask whether a difference is “genetic.” Saying a phenotypic difference among individuals is genetic often is implicitly taken as implying that it is immutable or unavoidable. However, even if we could attribute a some proportion of the difference in phenotypes between groups to polygenic scores, it would not lend support to the idea that this difference is immutable or “natural”. That is simply not how genetic variation works, as many phenotypes where genetics plays a role are modifiable.Without at least some working knowledge of causal mechanisms underlying the action of the genetic variation contributing to a trait, we may often not know how environment and culture shape the actions of these variants, nor how changes in these factors may modify any role played by genetics. Even if our tea polygenic scores were strongly predictive within and among populations, would cultural changes, e.g. a Europe-wide health food craze for drinking tea with dinner, stand these results on their head? Will taking tea with a meal moderate the role of caffeine-sensitivity SNPs; will exercise-conscious people now drink more tea? Will we know enough about the interaction of culture and genetics to predict this? If we do not, the statement that a difference in polygenic scores plays a role in explaining a difference in phenotypes among populations may often have little to say about how we as individuals or societies should view that difference. But will these critical subtleties be lost in the public’s understanding of results based on polygenic scores? Will such results be wrongly taken as supporting genetic determinism about human variation?

Pingback: Open Thread, 3/14/2018 – Gene Expression
Pingback: Links 1/19/19 | Mike the Mad Biologist
Pingback: Are GWAS studies of IQ/educational attainment problematic? « Statistical Modeling, Causal Inference, and Social Science
Pingback: Mining DNA for Disease Prediction: the polygenic risk score - Science in the News